Bu madde henüz onaylanmamıştır.

Turner Sendromu ( Yapay Zeka Tarafından Oluşturuldu.)
Ad(lar) | Turner Sendromu | ||||||||
|---|---|---|---|---|---|---|---|---|---|
Takip | Genetik danışmanlık, endokrin, kardiyoloji, nefroloji kontrolü | ||||||||
Komplikasyonlar | Hipertansiyon, infertilite, kardiyak sorunlar | ||||||||
Tedavi | Büyüme hormonu, östrojen replasmanı, multidisipliner takip | ||||||||
Tanı | Karyotip analizi | ||||||||
Belirtiler | Kısa boy, primer amenore, gonadal yetmezlik, lenfödem, kalp ve böbrek anomalileri | ||||||||
Sıklık | Kadınlarda 1/2,500 – 1/5,000 doğum | ||||||||
Genetik | Monozomi X (45,X) | ||||||||
Turner Sendromu, yalnızca kadın bireylerde gözlenen ve temel patogenetik mekanizması X kromozomunun tamamen ya da kısmen kaybına, yapısal anomalilerine veya mozaik dağılımına dayanan, fenotipik açıdan son derece geniş ve heterojen bir klinik spektrum sergileyen, büyüme, pubertal gelişim, üreme fonksiyonları, kardiyovasküler yapı, renal sistem, endokrin denge, nörokognitif süreçler ve psikososyal uyum gibi pek çok sistemi eş zamanlı olarak etkileyebilen, bu nedenle tanı, tedavi ve uzun dönem izlem süreçlerinde multidisipliner yaklaşımı zorunlu kılan kompleks ve kronik bir genetik hastalık olarak tanımlanmaktadır.
Turner sendromu, ilk kez 1938 yılında Henry Turner tarafından kısa boy, gonadal yetersizlik ve boyun derisinde katlantı gibi karakteristik bulgularla tanımlanmış olup, sitogenetik teknolojilerin gelişmesiyle birlikte 1950’li yıllarda hastalığın temelinde yatan kromozomal anöploidinin keşfedilmesi, bu sendromun patofizyolojisinin anlaşılmasında dönüm noktası olmuştur; ilerleyen yıllarda moleküler genetik alanındaki gelişmeler sayesinde X kromozomu üzerindeki belirli gen bölgelerinin kaybının klinik fenotip üzerindeki etkileri daha ayrıntılı biçimde ortaya konulmuş ve böylece Turner sendromunun yalnızca bir kromozom sayısı anomalisi değil, aynı zamanda gen dozajı dengesizliği ile ilişkili karmaşık bir biyolojik durum olduğu anlaşılmıştır.
Turner sendromunun genetik altyapısı incelendiğinde, hastalığın temelinde X kromozomunun tamamen yokluğu (45,X monozomisi), kısmi kaybı veya yapısal bozuklukları (izokromozom, ring kromozom, delesyonlar) ya da mozaik dağılımı (örneğin 45,X/46,XX veya 45,X/46,XY) gibi farklı sitogenetik varyantların yer aldığı görülmekte olup, bu varyasyonlar hastalığın klinik şiddetini ve bulguların çeşitliliğini doğrudan belirleyen en önemli faktörler arasında kabul edilmektedir.
X kromozomu üzerinde yer alan ve özellikle büyüme ile ilişkili olan pseudootozomal bölgede bulunan SHOX geninin haploinsufficiency durumu, Turner sendromunda gözlenen kısa boy gelişiminin moleküler temelini oluşturmaktadır; bu genin normalde iki kopya halinde bulunması gerekirken Turner sendromlu bireylerde tek kopya halinde bulunması, uzun kemiklerin epifiz plaklarında büyüme hızının azalmasına neden olmakta ve sonuç olarak belirgin boy kısalığı ortaya çıkmaktadır.
Bunun yanı sıra X kromozomu üzerinde bulunan ve over gelişimi ile ilişkili genlerin eksikliği, gonadal disgenez gelişimine yol açmakta ve bu durum pubertal gelişimin başlamasını engelleyerek infertiliteye neden olmaktadır; ayrıca X kromozomu üzerindeki bazı genlerin immün sistem regülasyonunda rol oynadığı ve bu genlerin eksikliğinin Turner sendromlu bireylerde otoimmün hastalıklara yatkınlığı artırdığı da bilinmektedir.
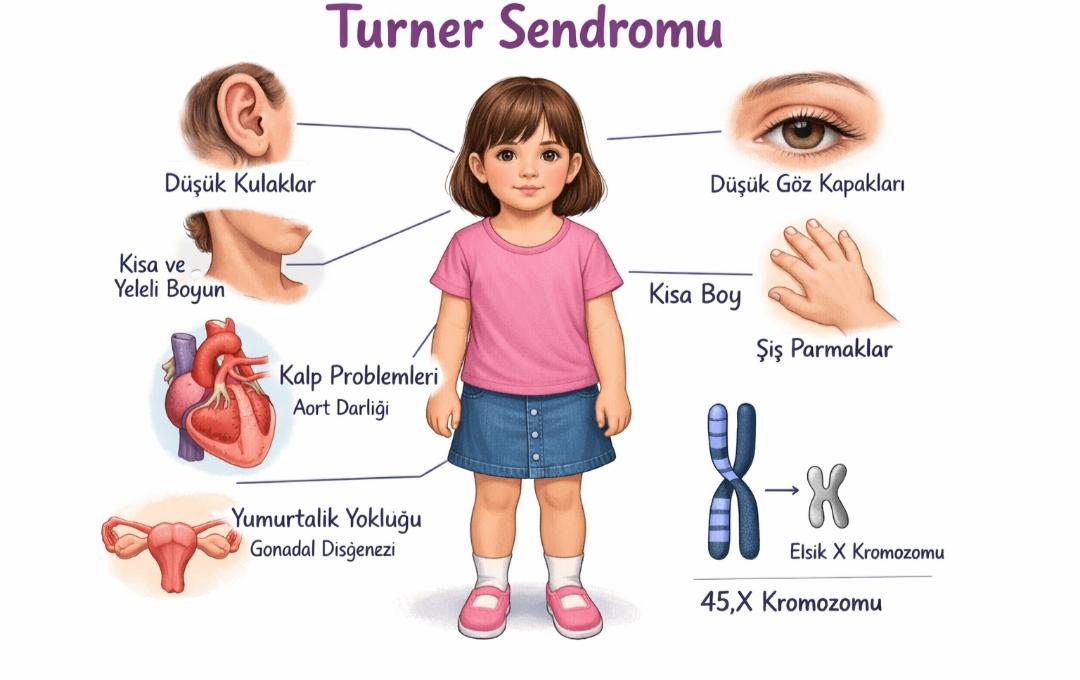
Turner Sendromu.(Yapay Zeka Tarafından Oluşturuldu.)
Turner sendromunun etkileri yalnızca kromozomal düzeyde sınırlı kalmamakta, aynı zamanda embriyonik gelişim sürecinde hücresel proliferasyon, migrasyon ve diferansiyasyon mekanizmalarını etkileyerek birçok organ sisteminin gelişimini sekteye uğratmaktadır; özellikle lenfatik sistem gelişiminde meydana gelen anomaliler, fetal dönemde boyun bölgesinde kistik higroma oluşumuna ve doğum sonrası dönemde boyun derisinde katlantı (webbed neck) görünümüne yol açmaktadır.
Ayrıca kardiyovasküler sistemin embriyonik gelişimi sırasında ortaya çıkan anomaliler, özellikle aort arkı ve kapak yapılarında bozukluklara neden olmakta ve bu durum ilerleyen yaşam dönemlerinde ciddi kardiyak komplikasyonların gelişmesine zemin hazırlamaktadır.
Turner sendromu yaklaşık olarak her 2000–2500 canlı kız doğumunda bir görülmekle birlikte, bu sendromun prenatal dönemde çok yüksek oranda spontan abortus ile sonuçlandığı bilinmektedir; yapılan çalışmalar, 45,X karyotipine sahip fetüslerin büyük çoğunluğunun gebeliğin erken dönemlerinde kaybedildiğini ve canlı doğumla sonuçlanan vakaların aslında tüm Turner sendromu olgularının yalnızca küçük bir kısmını temsil ettiğini göstermektedir.
Turner sendromunun klinik bulguları, bireyler arasında büyük değişkenlik göstermekle birlikte, genellikle doğumdan itibaren belirli fiziksel özellikler ve büyüme geriliği ile kendini göstermekte ve ergenlik döneminde pubertal gelişim yetersizliği ile daha belirgin hale gelmektedir.
Büyüme ve Somatik Özellikler: Turner sendromlu bireylerde büyüme geriliği genellikle erken çocukluk döneminde fark edilmekte olup, büyüme hızının yaşa göre düşük seyretmesi ve ergenlik döneminde beklenen büyüme atağının gerçekleşmemesi sonucunda erişkin boyun belirgin şekilde kısa kaldığı gözlemlenmektedir; bunun yanı sıra geniş göğüs kafesi, meme başlarının lateral yerleşimi, kısa ve geniş boyun, düşük posterior saç çizgisi, kubitus valgus ve lenfödem gibi karakteristik somatik bulgular sıklıkla dikkat çekmektedir.
Gonadal ve Endokrin Bulgular: Gonadal disgenez, Turner sendromunun en belirgin özelliklerinden biri olup, overlerin normal gelişim gösterememesi sonucu östrojen üretiminin yetersiz kalmasına ve bunun sonucunda pubertenin spontan olarak başlamamasına neden olmaktadır; bu durum primer amenore ile sonuçlanmakta ve hastaların büyük çoğunluğunda infertiliteye yol açmaktadır, ancak mozaik karyotipe sahip bazı bireylerde sınırlı da olsa spontan pubertal gelişim gözlenebilmektedir.
Kardiyovasküler Sistem Bulguları: Turner sendromlu bireylerde kardiyovasküler anomaliler, yaşam süresini belirleyen en önemli faktörlerden biri olup, en sık görülen anomaliler arasında biküspit aort kapağı ve aort koarktasyonu yer almakta, ayrıca bu bireylerde aort dilatasyonu ve diseksiyon riski normal popülasyona göre belirgin şekilde artmış bulunmaktadır; bu nedenle kardiyolojik değerlendirme ve düzenli takip hayati önem taşımaktadır.
Renal ve Ürogenital Bulgular: Renal sistem anomalileri Turner sendromunda oldukça sık görülmekte olup, özellikle at nalı böbrek, çift toplayıcı sistem ve renal malrotasyon gibi yapısal farklılıklar dikkat çekmektedir; bu anomaliler genellikle asemptomatik seyretmekle birlikte, bazı durumlarda idrar yolu enfeksiyonlarına ve böbrek fonksiyon bozukluklarına zemin hazırlayabilmektedir.
Nörokognitif ve Psikososyal Özellikler: Turner sendromlu bireylerin genel zeka düzeyleri çoğunlukla normal olmakla birlikte, özellikle uzaysal algı, matematiksel işlem yapma ve yürütücü işlevler gibi bilişsel alanlarda belirgin zorluklar yaşayabildikleri ve bu durumun akademik performans üzerinde olumsuz etkiler yaratabildiği bilinmektedir; ayrıca sosyal ilişkilerde çekingenlik, düşük özgüven, anksiyete ve depresyon gibi psikososyal sorunların daha sık görüldüğü ve bu nedenle psikolojik destek hizmetlerinin tedavi sürecine entegre edilmesinin önemli olduğu vurgulanmaktadır.
Turner sendromunun tanısı, klinik bulguların dikkatli değerlendirilmesi ve sitogenetik analizlerin yapılması ile konulmakta olup, karyotip analizi kesin tanı için altın standart olarak kabul edilmektedir; prenatal dönemde ise amniyosentez veya koryon villus örneklemesi ile kromozomal analiz yapılabilmekte ve böylece erken tanı mümkün hale gelmektedir.
Ayırıcı tanıda, kısa boy ve pubertal gecikme ile seyreden diğer endokrin ve genetik hastalıkların göz önünde bulundurulması gerekmekte olup, özellikle büyüme hormonu eksikliği ve diğer kromozomal anomaliler ile dikkatli bir şekilde ayrım yapılmalıdır.
Turner sendromunun tedavisi küratif olmamakla birlikte, hastalığın semptomlarını azaltmaya ve yaşam kalitesini artırmaya yönelik uzun süreli bir yönetim sürecini içermektedir; bu süreçte büyüme hormonu tedavisi ile boy uzamasının desteklenmesi, östrojen replasman tedavisi ile pubertal gelişimin sağlanması, kardiyovasküler ve renal sistemlerin düzenli olarak izlenmesi ve gerektiğinde cerrahi müdahalelerin uygulanması temel tedavi yaklaşımlarını oluşturmaktadır.
Bunun yanı sıra işitme sorunlarının erken dönemde tespit edilerek uygun şekilde tedavi edilmesi, tiroid fonksiyonlarının düzenli olarak izlenmesi ve metabolik risk faktörlerinin kontrol altına alınması da uzun dönem prognoz açısından büyük önem taşımaktadır.
Akçan, A. Barış. “Turner Sendromu.” Konuralp Medical Journal 5, no. 2 (2013): 53–61. Erişim Tarihi 6 Nisan 2026.
Kendir Demirkol, Yasemin. “Turner Sendromu.” İçinde Çocuk Genetik Uygulamalarında Sık Görülen Hastalıkların Takip ve Tedavisi, editör E. Mıhçı, 9–16. Ankara: Türkiye Klinikleri, 2021. Erişim Tarihi 6 Nisan 2026.
https://www.turkiyeklinikleri.com/article/en-turner-sendromu-95592.html
Yalçın, Sıddıka Songül, Tuba Çelen Yoldaş, ve G. Eda Ütine. “Turner Sendromunun Nörogelişimsel ve Psikososyal Sorunlarında Olgu Yönetim Rehberi.” Turkish Journal of Pediatric Disease 12, no. 1 (2018): 62–68. Erişim Tarihi 6 Nisan 2026.
Ünal, Edip, ve Yusuf Kenan Haspolat. “Turner Sendromlu Olguların Değerlendirilmesi.” Dicle Üniversitesi Tıp Fakültesi Dergisi (2020). Erişim Tarihi 6 Nisan 2026.
Turner Sendromu ( Yapay Zeka Tarafından Oluşturuldu.)
Ad(lar) | Turner Sendromu | ||||||||
|---|---|---|---|---|---|---|---|---|---|
Takip | Genetik danışmanlık, endokrin, kardiyoloji, nefroloji kontrolü | ||||||||
Komplikasyonlar | Hipertansiyon, infertilite, kardiyak sorunlar | ||||||||
Tedavi | Büyüme hormonu, östrojen replasmanı, multidisipliner takip | ||||||||
Tanı | Karyotip analizi | ||||||||
Belirtiler | Kısa boy, primer amenore, gonadal yetmezlik, lenfödem, kalp ve böbrek anomalileri | ||||||||
Sıklık | Kadınlarda 1/2,500 – 1/5,000 doğum | ||||||||
Genetik | Monozomi X (45,X) | ||||||||
Henüz Tartışma Girilmemiştir
"Turner Sendromu" maddesi için tartışma başlatın
TARİHÇE
GENETİK VE MOLEKÜLER TEMELLER
EMBRİYOLOJİK VE GELİŞİMSEL SÜREÇLER
EPİDEMİYOLOJİ VE PRENATAL KAYIPLAR
KLİNİK BULGULAR
TANI YÖNTEMLERİ VE AYIRICI TANI
TEDAVİ, İZLEM VE YAŞAM BOYU YÖNETİM